DNA methylation is an epigenetic mechanism that occurs by the addition of a methyl (CH3) group to DNA, thereby often modifying the function of the genes and affecting gene expression. The most widely characterized DNA methylation process is the covalent addition of the methyl group at the 5-carbon of the cytosine ring resulting in 5-methylcytosine (5-mC), also informally known as the “fifth base” of DNA. These methyl groups project into the major groove of DNA and inhibit transcription.
In human DNA, 5-methylcytosine is found in approximately 1.5% of genomic DNA.1 In somatic cells, 5-mC occurs almost exclusively in the context of paired symmetrical methylation of a CpG site, in which a cytosine nucleotide is located next to a guanidine nucleotide. An exception to this is seen in embryonic stem (ES) cells, where a substantial amount of 5-mC is also observed in non-CpG contexts. In the bulk of genomic DNA, most CpG sites are heavily methylated while CpG islands (sites of CpG clusters) in germ-line tissues and located near promoters of normal somatic cells, remain unmethylated, thus allowing gene expression to occur. When a CpG island in the promoter region of a gene is methylated, expression of the gene is repressed (it is turned off).
The addition of methyl groups is controlled at several different levels in cells and is carried out by a family of enzymes called DNA methyltransferases (DNMTs). Three DNMTs (DNMT1, DNMT3a and DNMT3b) are required for establishment and maintenance of DNA methylation patterns. Two additional enzymes (DNMT2 and DNMT3L) may also have more specialized but related functions. DNMT1 appears to be responsible for the maintenance of established patterns of DNA methylation, while DNMT3a and 3b seem to mediate establishment of new or de novo DNA methylation patterns. Diseased cells such as cancer cells may be different in that DNMT1 alone is not responsible for maintaining normal gene hypermethylation (an increase in global DNA methylation) and both DNMTs 1 and 3b may cooperate for this function.
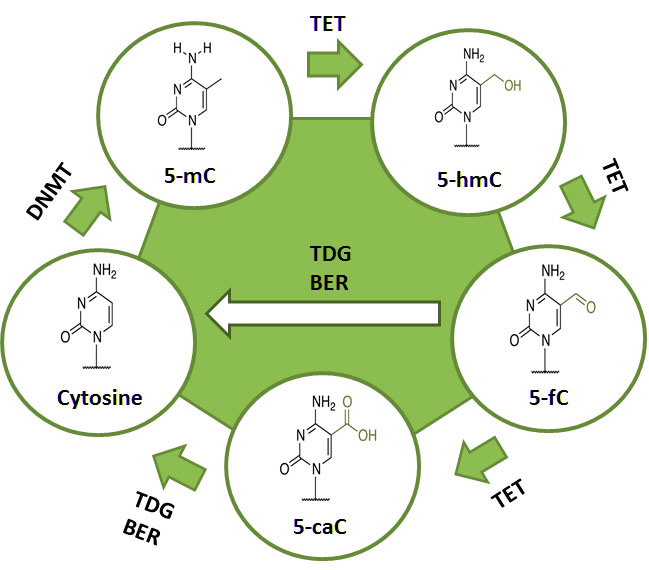
DNA demethylation is the removal of a methyl group from DNA. This mechanism is equally as important and coupled with DNA methylation. The demethylation process is necessary for epigenetic reprogramming of genes and is also directly involved in many important disease mechanisms such as tumor progression. Demethylation of DNA can either be passive or active, or a combination of both. Passive DNA demethylation usually takes place on newly synthesized DNA strands via DNMT1 during replication rounds. Active DNA demethylation mainly occurs by the removal of 5-methylcytosine via the sequential modification of cytosine bases that have been converted by TET enzyme-mediated oxidation. The ten-eleven translocation (TET) family of 5-mC hydroxylases includes TET1, TET2 and TET3. These proteins may promote DNA demethylation by binding to CpG rich regions to prevent unwanted DNA methyltransferase activity, and by converting 5-mC to 5-hmC, 5-hmC to 5-fC (5-formylcytosine), and 5-fC to 5-caC (5-carboxylcytosine) through hydroxylase activity. The TET proteins have been shown to function in transcriptional activation and repression (TET1), tumor suppression (TET2), and DNA methylation reprogramming processes (TET3).
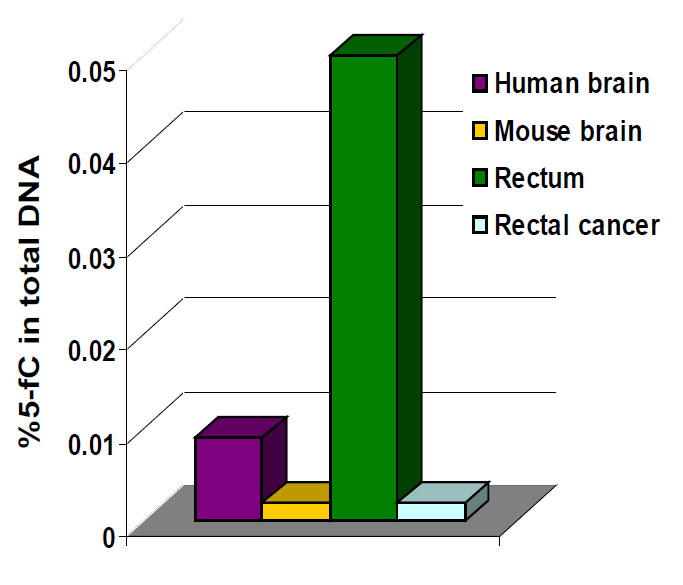
The biological importance of 5-mC as a major epigenetic modification in phenotype and gene expression has been widely recognized. For example DNA hypomethylation, the decrease in global DNA methylation, is likely caused by methyl-deficiency due to a variety of environmental influences and has been proposed as a molecular marker in multiple biological processes such as cancer. The quantification of 5-mC content or global methylation in diseased or environmentally impacted cells could provide useful information for detection and analysis of disease. Furthermore, the detection of the DNA demethylation intermediate 5-fC in various tissues and cells may also be used as a marker to indicate active DNA demethylation. 5-fC can also be directly excised by thymine DNA glycosylase (TDG) to allow subsequent base excision repair (BER) processing which converts modified cytosine back to its unmodified state.
Differentially methylated regions (DMRs) are areas of DNA that have significantly different methylation status between multiple samples. Researchers will often perform genome-wide methylation profiling to identify DMRs between treated or untreated samples, revealing functional regions that may be involved in gene transcriptional regulation. There can be DMRs specific to tissues, cells, individuals, and so on. Differentially methylated regions may also be used as biomarkers or potential targets of epigenetic therapy.
Continue to the next page to learn about DNA methylation tools of the trade.